Comparison of RNA-Seq Aligners for Low-Abundance Transcripts
Video
Team Information
Team Members
Theodore Nelson, Undergraduate Student in Computer Science, Columbia College
Faculty Advisor: Thomas Postler, Assistant Professor of Microbiology & Immunology, Columbia University Irving Medical Center
Abstract
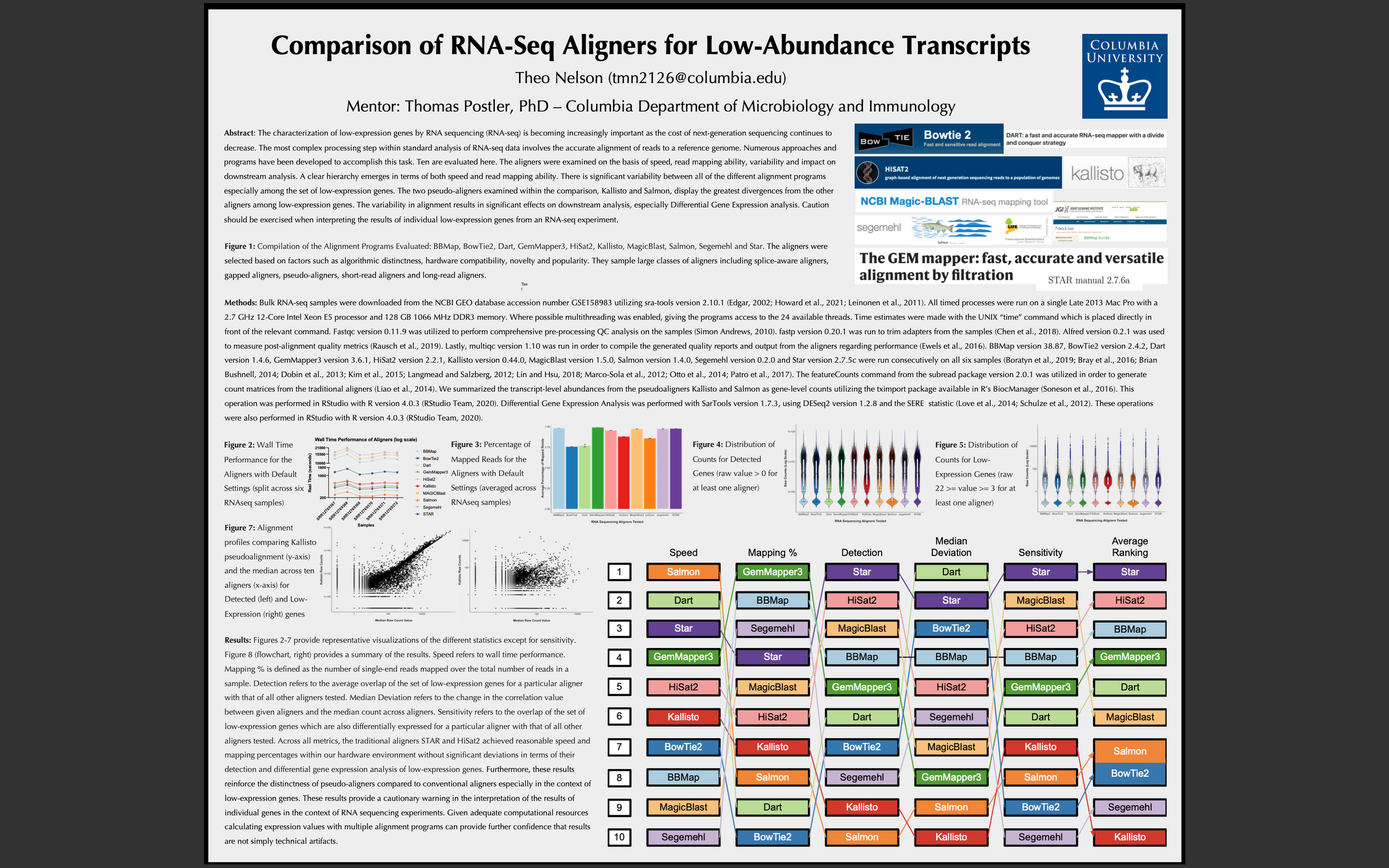
The characterization of low-expression genes by RNA sequencing (RNA-seq) is becoming increasingly important as the cost of next-generation sequencing continues to decrease. The most complex processing step within standard analysis of RNA-seq data involves the accurate alignment of reads to a reference genome. Numerous approaches and programs have been developed to accomplish this task. Ten are evaluated here. The aligners were examined on the basis of speed, read mapping ability, variability and impact on downstream analysis. A clear hierarchy emerges in terms of both speed and read mapping ability. There is significant variability between all of the different alignment programs especially among the set of low-expression genes. The two pseudo-aligners examined within the comparison, Kallisto and Salmon, display the greatest divergences from the other aligners among low-expression genes. The variability in alignment results in significant effects on downstream analysis, especially Differential Gene Expression analysis. Caution should be exercised when interpreting the results of individual low-expression genes from an RNA-seq experiment.
Team Lead Contact
Theodore Nelson: tmn2126@columbia.edu